Перспективный метод определения белковых структур набирает обороты
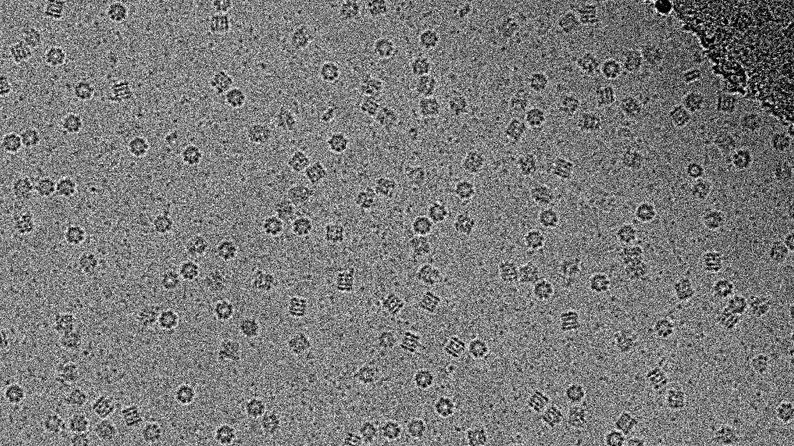
Более 10 000 молекул набралось в базе белковых структур, определенных с помощью электронной микроскопии (Electron Microscopy Data Bank, EMDB), 5 февраля сообщает EMDataResourсe.
Ученым всего мира понадобилось 18 лет, чтобы достичь рубежа в 10 000 белков, визуализированных с помощью электронной микроскопии. Для сравнения, база белковых структур (Protein Data Bank, PDB), определенных рентгеноструктурным анализом (РСА) и ЯМР-спектроскопией, начала пополняться с 1971 года и перевалила через эту отметку только 29 лет спустя.
EMDB запущена в 2002 году Европейским институтом биоинформатики (EBI). Большинство загруженных в эту базу белковых структур определены методом криоэлектронной микроскопии (крио-ЭМ).
Крио-ЭМ подразумевает быстрое замораживание образца в жидком азоте и пропускание сквозь него пучка электронов. Компьютерная обработка полученных изображений позволяет реконструировать поверхностный ландшафт и трехмерную структуру исследуемого белка.
Поначалу метод давал настолько размытые изображения, что его иронически называли «блобологией» от английского слова blob («капля»). Все изменилось с внедрением более чувствительных электронных микроскопов и усовершенствованных алгоритмов обработки в 2012–2013 гг., хотя отдельные белковые структуры публиковались и раньше.
На сегодняшний день крио-ЭМ позволяет определять пространственную структуру белков с разрешением 2–4 Å (один ангстрем (Å) равен 10-10 м — прим. ИА Красная Весна). Это дает возможность устанавливать координаты отдельных атомов, за исключением водорода.
В отличие от РСА, где 3D-структуру биомолекул определяют по дифракции рентгеновских лучей на кристаллической решетке, крио-ЭМ не требует кристаллизации образцов. Многие исследователи проводят годы в попытках закристаллизовать тот или иной белок для РСА. Даже когда им это удается, в результирующей структуре могут отсутствовать относительно подвижные участки биомолекул, а сама она не всегда соответствует той, которую белок имеет в клетке.
Крио-ЭМ также не ограничена размером белков и не требует того, чтобы они были растворимы. Это дает методу преимущество и над спектроскопией ядерного магнитного резонанса (ЯМР), которая позволяет исследовать только небольшие биомолекулы в растворе.
В 2017 году швейцарец Жан Дюбоше, британец Ричард Хендерсон и немец Йоахим Франк удостоились Нобелевской премии по химии за внедрение крио-ЭМ в структурную биологию. В 1990 году Хендерсон опубликовал структуру белка бактериородопсина с разрешением 3,5 Å (для сравнения, длина отдельной углерод-углеродной связи — 1,1–1,5 Å).
При сравнении перспектив РСА и крио-ЭМ примечательно сопоставить, сколько белковых структур год от года расшифровывается каждым из этих методов. Применительно к РСА их количество вышло на плато к 2016 году и с тех пор не росло, зато в случае крио-ЭМ оно увеличивается примерно с того же момента взрывными темпами.
Сегодня с помощью крио-ЭМ определяется каждая четвертая белковая структура, публикуемая в базе данных PDB. Исходя из наметившихся тенденций и из особенностей обоих методов, можно предположить, что в ближайшем будущем ниша РСА значительно сузится, хотя он и останется актуальным для тех случаев, где требуется сверхвысокое разрешение. В то же время крио-ЭМ станет незаменимой при изучении структурно подвижных белков в различных конформационных состояниях, переходы между которыми не фиксируются в РСА.














